
The Internet loves cats, so I figure Cloud will garner me more views than all my other blog posts combined
When my wife gifted me a Basepaws Whole Genome Sequencing kit for our beloved cat Cloud last Christmas, I was pretty stoked. Not just because it’s exceptionally cool that this technology exists and is commercially viable, but also because I am so accustomed to analyzing human genomic data that, as a bioinformatician, I can appreciate just how challenging it is to discover and report on genuinely meaningful findings from a relatively lesser-studied genome such as that of Felis catus.
I was curious to know to what extent this company could provide interesting or actionable info on my pet, as well as to what extent I could glean something useful from the data myself after running analyses on the raw sequencing data. This is no simple task — in order to complete genomic analyses cost-effectively, many complex tools need to be pipelined together, and the execution of each step must be orchestrated to run on machines of various sizes and capabilities. Who am I to deny such a challenge for the sake of my cute critter?
Before I discuss how I leveraged the latest version of Cloud Composer (GCP’s fully managed Apache Airflow service) to simplify the automated execution and visualization of several complex genomic analysis steps, as well as how I leveraged Claude 3.5 Sonnet on AWS Bedrock to analyze the results, I want to start by highlighting how impressed I was with my Basepaws-generated PDF report.
Clocking in at 68 pages, there’s more to it than I have time to cover in this article. However, in short, the report clearly communicates for your pet:
- Breed group data (a sample of this is shown below)
- The presence/absence of genetic health markers / genetic disorders
- A multitude of interesting trait markers
- And, to my surprise, an oral health report based on the oral microbiome
The oral health report in particular was pretty cool, as it confirmed some already-known issues (e.g., ‘Medium’ risk for periodontal disease) and confirmed that actions taken to mitigate other issues have been effective (e.g., ‘Low’ risk for tooth resorption, a likely partially genetic-in-origin issue he has had for years that my vets have successfully addressed).
It’s difficult enough to analyze the relatively poorly annotated cat genome, but to also sequence the oral microbiome and provide meaningful, actionable analytics on that is a real accomplishment. Bravo to the team of scientists that made this happen.

I’ve considered getting a Maine Coon as my next cat — I wouldn’t have guessed I already (sorta) had one!
With that said, let’s explore what you could do with the raw whole genome data provided to you by this company alongside their report.
Linked below is a repo associated with this blog that leverages the recently released v3 of Cloud Composer and a few other cutting-edge GCP services—all spun up with the provided Terraform—to help you fully automate, scale, and visualize the execution of the complex web of tools required to perform secondary and tertiary analysis on cat whole genome data.
https://github.com/doit-mattporter/genomics-workflow-orchestration
Let’s begin our discussion on how to run cat genomic analytics within GCP by first gaining a basic understanding of what this repo will be executing.
For those unfamiliar with bioinformatics: Generally speaking, there are three major pipeline stages that represent how you will analyze genomic data, regardless of the organism you’re working with. Primary analysis will be taken care of by the time you get your sequencing data from Basepaws, while secondary and tertiary analyses are run with the help of the code base I’ve provided. These three stages are described below:
- Primary analysis. This step takes place at the DNA sequencing facility and encompasses converting raw signal data (light data) from sequencing machines into sequences of DNA base pairs (sequences of A/C/G/T). It also includes QC checks, such as removing low-confidence sequence calls.
Due to technical limitations, it is impossible to sequence a DNA strand from beginning to end in one massive, continuous reading. DNA strands must instead be chopped up into millions of short pieces that are only then capable of being sequenced. The output of primary analysis will thus be millions — if not billions — of short DNA fragments, each typically 100 to 300 base pairs (bps) in length. This raw, fragmented sequencing data will be provided to you in FASTQ format, essentially a plain text format consisting of strings of ACGT base pair values for these millions of short sequences. This is what BasePaws sends you and what this blog/associated repo will begin working with. 2. Secondary analysis. This stage is like putting together a giant puzzle of your DNA, where the solution is always a little different from what’s shown on the box. There are 2 major components to secondary analysis:
a) ‘Sequence Alignment,’ aka DNA Puzzle Assembly: Those millions of short DNA fragments from primary analysis are pieced back together — with a great deal of computing power — to yield the original, fully intact cat genome those fragments originated from.
b) ‘Variant Calling’, aka Spot the Differences: Once your cat’s genome has been reassembled, it must be compared against the ‘reference’ genome that the scientific community has determined is the gold standard for what a typical cat genome looks like. Your cat will deviate from this ‘reference’ standard due to their genetic uniqueness; this step involves looking for those genetic differences, places where your cat’s DNA differs from ‘typical’ cat DNA. These differences are called ‘variants’ or ‘mutations’. Not all mutations discovered during variant calling are real — many variants are mistakes, for complicated reasons. This step also involves QC, making sure the variants are real. 3. Tertiary analysis. Often the trickiest and most crucial part, this represents discovering what the unique list of DNA differences found from secondary analysis actually means. Tertiary analysis represents the presentation of tangible/meaningful findings. Do some of these variants have known associations with health conditions? How will my cat respond to various pharmaceutical drug options based on their list of mutations? Is this particular mutation responsible for their coat color? Does this set of mutations mean my cat is likely of Maine Coon ancestry? Tertiary analysis encompasses the ability to answer questions of this nature.
Now that you have a good enough idea of the work to be done, let’s get to it! Once you have received your cat's whole genome data from BasePaws — or if you just want to follow along and see what’s possible with Cloud Composer and Claude using this data — you can proceed with the following.
The repo’s README describes how you should begin by provisioning the required cloud infrastructure with aterraform applyoperation. Note that some resources, notably Cloud Composer, will run 24/7 until terminated, so be mindful of your cloud spend and be sure to terminate unused resources. The following key resources will be spun up:
Google Composer Environment
- Composer 3 environment with Apache Airflow 2
- Configures an Airflow DAG for cat genomic data processing. Note that this DAG will temporarily provision c4-standard-96 and c4-standard-8 Compute Engine instances as required by various pipeline steps.
Google Cloud Storage Buckets
genomic_inputs,genomic_reference,genomic_outputs, andcloud_functionfor storing inputs (FASTQ files), reference genomes, outputs (VCFs and annotation files), and Cloud Function code
Google Cloud Function v2
genomic-dag-kickoffv2 function to trigger genomic analytics DAG- Event-driven by GCS object finalization. This function checks for a
ready.txtfile, then uses that file’s list of FASTQ bucket locations to kick off the DAG to process those FASTQs.
Google Compute Engine instance
grabbing-reference-genomeis a c4-standard-96 instance that downloads two cat reference genomes from NCBI, indexes these references with GATK, and then uploads these processed reference genomes to thegenomic_referencebucket for later use by the DAG.- Note that this resource only needs to be run once and will self-terminate once its work has been completed.
Once those resources are up and running from terraform apply, you simply do the following to kick off cat genome analytics:
- Upload BasePaws FASTQs to the
genomic_inputs_<random_id>bucket - Create a
ready.txtfile that contains the GCS URIs for those FASTQs and upload that file to the same bucket.
Upload of the ready.txt file will trigger the genomic-dag-kickoff Cloud Function. This function will start the execution of two DAG instances on Cloud Composer, each one orchestrating the execution of secondary and tertiary analysis against one of the two latest versions of the cat genome: Felis_catus_9.0 and Felis_catus_Fca126_mat1.0.
The DAG executes a complex orchestration of bioinformatics tools that ultimately gives us:
- VCF files, or Variant Call Files. This is a list of all discovered mutations.
- Annotation files produced by SnpEff. SnpEff provides in silico annotations for each mutation located within a protein-coding gene with the likelihood that mutation will result in a ‘Low,’ ‘Moderate,’ or ‘High’ functional impact on the protein encoded by that gene.
At a high level, the DAG is composed of the following Task Groups, with ‘alignment’ and ‘variant calling’ Task Groups corresponding to Secondary Analysis, and the ‘annotation’ Task Group representing Tertiary Analysis:
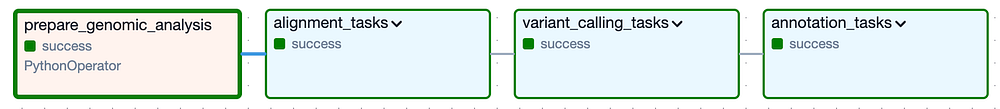
Cloud Composer’s DAG for executing secondary and tertiary analysis on a particular cat genome version
Expanding each of the Task Groups (shown below) reveals the many individual tasks required to run this pipeline of tools and then validate that the expected output files were created. Note that I’ve included logic to skip each Task Group entirely if its output files are already present in the output bucket from a previously successful run.
Without the help of Apache Airflow and GCP’s fully managed version of this open-source tool, pipelining these tools and verifying their successful execution (or previously successful execution) at each step would be substantially more challenging and time-consuming to implement. Not only would it be challenging to orchestrate these steps but also to retrieve and store their logs, visualize real-time pipeline progress, and report on any issues.
Composer makes it easy to view progress, create pipelines with branching logic paths, and ensure errors are captured and their logs presented in an easy-to-parse UI. For these reasons and many others — such as Composer’s ability to cost-effectively scale compute resources in and out when used in enterprise-scale operations— I consider workflow orchestration tools like Composer mission-critical to building scalable data pipelines.
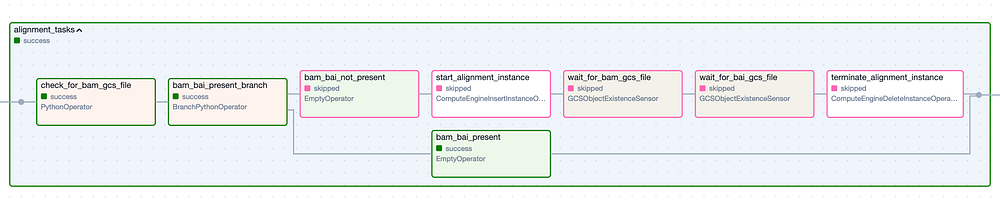
The ‘Alignment’ Task Group. Green border steps represent successful executions, while pink border steps were skipped.
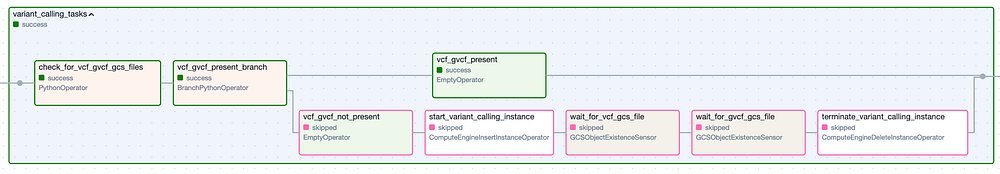
The ‘Variant Calling’ Task Group. Green border steps represent successful executions, while pink border steps were skipped.

The ‘Annotation’ Task Group. Green border steps represent successful executions, while pink border steps were skipped.
Once these all run to completion, you will see VCFs and annotation files dumped into the genomic-outputs-<random_id> GCS bucket. VCFs contain a list of all mutations discovered, while the annotation files contain the details of SnpEff’s in silico predicted effects of those mutations on the proteins that genes encode for.
We’re interested specifically in how SnpEff believes protein-coding genes are likely to be impacted.
This is where my appreciation for the BasePaws service really began to kick in. With human genomics, there is an astounding wealth of open-source variant annotation data sources you can draw from to gain a deep clinical understanding of just about any mutation that occurs within a protein-coding gene. By contrast, in the realm of cat genomics, I failed to find any open-source annotation database, not even a database for mutations with clear, known clinical outcomes. As I am not eager to scrape academic papers to build my own database, I was only left with implementing SnpEff in my Cloud Composer DAG’s annotation pipeline step, which will at least give us in silico functional effect predictions to draw upon. BasePaws must have put in a great deal of effort building annotation databases internally in order to offer the kinds of analytics I saw in the report they provided.
Alas, determined to get some use out of SnpEff’s effect predictions, I turned to LLMs, specifically Claude 3.5 Sonnet running on AWS Bedrock.
I began by running the following bash commands on the annotation file SnpEff produces. These simple commands filter the 13.5 million annotated mutations down to just 366 that are more likely to be ‘interesting’ — those that are:
- Located within a protein-coding region
- More likely to be legitimate variants vs. a mistaken variant call
- Have an identified human homolog (an LLM will be more likely to understand the potential impact if it knows its human protein equivalent), and
- Have a ‘HIGH’ predicted impact to the functional effect of a protein
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep "\tHIGH\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep -v "frameshift" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv |
grep -v "intron" |
awk '$11 !~ /^ENSFCAG000/' > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
I pasted the contents of that filtered file with 366 ‘HIGH’ impact annotated variants into Claude, along with the following prompt:
Analyze the following SnpEff genetic annotations for Felis catus 9.0. When referencing a gene, make sure to mention how many mutations were discovered in that gene and do not hallucinate mutations for genes that are not provided in the annotation file pasted below. Please provide:
1. A summary of the most significant findings
2. Highlight any mutations in genes associated with known health conditions, phenotypic traits, or important biological processes.
3. Explain the potential functional implications of the top 5–10 most interesting mutations, considering the gene’s function and the type of mutation.
4. Identify any patterns, clusters of mutations, or impacted biological/functional pathways that might be noteworthy.
5. Suggest any potential health concerns or traits that might be affected based on these mutations, but clarify that this is speculative without clinical correlation.
Please use scientific terminology where appropriate, but also provide layman explanations to ensure understanding. If you need any clarification or additional information about the subject’s phenotype or health history to improve your analysis, please ask.
The cat has an all white coat, is 11 years old, and has no known health issues other than feline tooth resorption. The cat has no known neurological or immune system disabilities (aside from the possible link between immune system function and feline tooth resorption).
This gave the following results, which I thought were pretty interesting as Claude does suggest a possible genetic predisposition for feline tooth readsorption based on some of the genes with high-impact mutations. With an all-white coat, the possibility that he might also be at a higher risk for cancer due to a DNA repair protein’s functionality possibly being knocked out is good to be aware of, as it does mean I should limit his exposure time to direct sunlight.
Claude’s other findings are quite speculative, however, as I have not observed any of the reported serious issues with muscle function or metabolism. Given there are no well-organized, publicly available annotation datasets, the function of cat genes is largely inferred via homology to their equivalent human genes. I therefore expect LLMs to respond with human-centric, broad functionality inferences, leading to the overly speculative results Claude has output.
Thank you for providing this detailed genetic information about your 11-year-old white cat with feline tooth resorption. I’ll analyze the high-impact mutations based on the SnpEff annotations you’ve shared. Let’s break this down into the requested sections:
1. Summary of Most Significant Findings:
The genetic data reveals a substantial number of high-impact mutations across various genes. These mutations include stop-gained, start-lost, and stop-lost variants, which can significantly alter protein function. In total, there are over 200 high-impact mutations identified across numerous genes.
2. Mutations in Genes Associated with Known Conditions or Traits:
a) LRRC45 (1 mutation): Associated with ciliary function, which can affect various physiological processes.
b) PNKP (1 mutation): Involved in DNA repair, potentially impacting cancer susceptibility and neurological function.
c) ATP2A1 (1 mutation): Critical for muscle function, particularly in fast-twitch muscle fibers.
d) COQ7 (1 mutation): Important for coenzyme Q10 biosynthesis, affecting energy production in cells.
e) ABCC6 (1 mutation): Associated with connective tissue disorders in humans.
f) CRP (1 mutation): Involved in immune response and inflammation.
3. Potential Functional Implications of Top Mutations:
a) PNKP (c.1150C>T, p.Gln384*): This stop-gained mutation could lead to a truncated protein, potentially impairing DNA repair mechanisms. This might increase susceptibility to DNA damage and theoretically could affect neurological function or cancer risk.
b) ATP2A1 (c.3149A>G, p.Ter1050Trpext*?): This stop-lost mutation might result in an extended protein. ATP2A1 is crucial for muscle contraction, and alterations could potentially affect muscle function.
c) COQ7 (c.817T>A, p.Ter273Lysext*?): Another stop-lost mutation, this could lead to an extended protein. COQ7 is vital for coenzyme Q10 production, which is essential for cellular energy production. Alterations might affect overall energy metabolism.
d) ABCC6 (c.4246C>T, p.Arg1416*): This stop-gained mutation could result in a truncated protein. In humans, ABCC6 mutations are associated with pseudoxanthoma elasticum, a condition affecting connective tissue.
e) CRP (c.592T>C, p.Ter198Glnext*?): This stop-lost mutation in the C-reactive protein gene could affect inflammatory responses and immune function.
4. Patterns and Noteworthy Observations:
There appears to be a cluster of mutations affecting genes involved in muscle function, energy metabolism, and immune response. Several mutations are found in genes related to DNA repair and cellular maintenance processes.
5. Potential Health Concerns or Traits (Speculative):
Based on these mutations, there might be potential concerns related to:
\* Muscle function and metabolism (ATP2A1, COQ7)
\* Immune system regulation (CRP)
\* DNA repair and potential cancer susceptibility (PNKP)
\* Connective tissue health (ABCC6)
However, it’s crucial to emphasize that the presence of these mutations doesn’t necessarily mean the cat will develop related conditions. Many factors, including environmental influences and other genetic modifiers, play a role in gene expression and disease development.
It’s interesting to note that despite these mutations, your cat has lived to 11 years with no major health issues reported aside from feline tooth resorption. This underscores the complex relationship between genotype and phenotype.
Given the cat’s history of feline tooth resorption, the mutations in genes related to immune function (like CRP) and connective tissue (like ABCC6) might be of particular interest, as these systems are thought to play a role in this condition. However, a direct link cannot be established without further research.
Next, I decided to look into what genes are known to be associated with a white coat color in cats, then set about filtering for all SnpEff-annotated mutations within these genes regardless of SnpEff’s low/medium/high impact effect prediction. This was accomplished by filtering the annotation file for mutations in the KIT and KITLG genes:
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
grep -E "\t(LOW|MODERATE)\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv | grep -E '\tKIT\t|\tKITLG\t' >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
Reusing the same prompt, Claude surmises that the missense and intron splice site mutations are likely responsible for Cloud’s white coat color:
Thank you for providing the genetic annotations for Felis catus 9.0. I’ll analyze this data and provide the requested information. Let’s break down the findings:
1. Summary of the most significant findings:
The annotations reveal multiple mutations in two important genes: KIT and KITLG. These genes are part of the KIT/KITLG signaling pathway, which plays crucial roles in various biological processes, including coat color development, hematopoiesis, and gametogenesis.
KIT gene: 9 mutations (7 synonymous variants, 2 splice region variants)
KITLG gene: 2 mutations (1 missense variant, 1 synonymous variant)
The most significant finding is the missense variant in the KITLG gene, which could potentially affect protein function.
2. Mutations associated with known health conditions, phenotypic traits, or important biological processes:
The KIT and KITLG genes are associated with coat color in cats, particularly white spotting patterns. The all-white coat of this cat might be related to these mutations, especially considering the number of variants in the KIT gene.
3. Potential functional implications of the top 5–10 most interesting mutations:
a) KITLG missense variant (c.731G>A, p.Ser244Asn): This mutation changes a serine to an asparagine at position 244 in the KITLG protein. This could potentially affect the protein’s function, as it alters the amino acid sequence. KITLG is crucial for the survival and proliferation of melanocytes, which produce pigment. This mutation might contribute to the cat’s all-white coat.
b) KIT splice region variants (c.2491–3C>T, c.1353–5T>C, c.1122–4_1122–3delGT): These mutations occur in splice regions, which are important for proper mRNA processing. Although they’re classified as low impact, they could potentially affect the splicing of the KIT gene, leading to altered protein production or function. The KIT protein is a receptor tyrosine kinase involved in various developmental processes, including melanocyte development.
c) KIT synonymous variants: While synonymous variants don’t change the amino acid sequence, they can still impact gene expression through various mechanisms such as altering mRNA stability or translation efficiency. The high number of synonymous variants in the KIT gene (7 in total) might collectively influence its expression or function.
4. Patterns, clusters of mutations, or impacted biological pathways:
The mutations are clustered in the KIT/KITLG signaling pathway, which is crucial for melanocyte development and function. This pathway is also involved in hematopoiesis (blood cell formation) and gametogenesis (production of reproductive cells). The concentration of mutations in these genes suggests a potential impact on pigmentation and possibly other related processes.
5. Potential health concerns or traits that might be affected (speculative):
a) Coat color: The all-white coat of the cat is likely related to these mutations, particularly in the KIT and KITLG genes.
b) Hearing: Some all-white cats with KIT mutations are prone to congenital deafness. While no specific deafness-associated mutations are present here, it might be worth monitoring the cat’s hearing.
c) Immune function: The KIT/KITLG pathway plays a role in hematopoiesis, including the development of certain immune cells. While the cat has no known immune system disabilities, these mutations could potentially influence immune function subtly.
d) Tooth resorption: Although not directly linked to KIT/KITLG in current literature, the pathway’s involvement in various developmental processes might indirectly contribute to dental issues. This is highly speculative and would require further research to establish any potential connection.
e) Fertility: Given the role of KIT/KITLG in gametogenesis, there could be potential effects on fertility. However, this is speculative and would only be relevant if the cat were to be bred.
It’s important to note that while these genetic findings are interesting, their clinical significance can only be determined through correlation with the cat’s actual health status and further studies. The cat’s current good health at 11 years old suggests that many of these mutations may not have severe impacts.
My takeaway from all of this is that it is possible to glean some pretty cool findings from your cat’s genomic data, and obtaining that genomic data is made substantially more feasible, reliable, and scalable with the help of cloud services that orchestrate data pipelines such as Cloud Composer.
However, much remains to be done on the publicly funded research side of things. Inferring cat gene functionality from their homology to human genes will only enable meaningful analytics to a limiting extent. When veterinary research institutions begin publishing genomic annotation databases for cats and dogs, I believe we’ll start to see LLM-powered analytics on genomic data truly shine. Until then, I’m left impressed with the scientific work that has been undertaken by the Basepaws team.
And there you have it! We’ve journeyed through the fascinating frontier of feline genomics, from BasePaws’ impressive report to our own DIY analysis powered by cutting-edge cloud computing services and generative AI. While orchestrating a symphony of genomic tools can be as complex as herding cats, services like Cloud Composer make it much easier.